- Quality and Scientific Regulatory Services for Global Bio-/Pharmaceutical Industry
Biosimilars & Vaccines Regulatory Support
Biosimilars & Vaccines Regulatory Support
Our Expertise in Biosimilars & Vaccines
- Skilled resources with core biopharmaceutical industry experience, having in-depth understanding of biologics (biosimilars and vaccine) development processes and regulatory requirements at different phases of product life cycle.
- Technical competencies to support regulatory activities related to different recombinant expression systems and mammalian cell lines (E.Coli, Yeast rCHO etc.) for biosimilar product development and various platform technologies (egg based, cell based, recombinant) for vaccine development.
- Specialized team for regulatory CMC authoring, dossier compilation, review and submissions, addressing deficiency responses from various Health Authorities.
- Expertise in generating biosimilar and vaccine intelligence reports.
- Strong connect with regulatory and marketing experts across BRICS-TM countries, ACCESS countries, Asia, Africa (including MENA), GCC and Latin America.

Where can Metina support?
- Development phase (CMC)
- Non-clinical Phase
- Phase I
- Phase II (for vaccines)
- Phase III
- Marketing authorization
Development phase (CMC)
- Regulatory guidance on upstream and downstream process development (DS) and product development (DS & DP) for biosimilars & vaccines.
- Review and advice for establishing biosimilarity – CMC, analytical comparability protocol (physicochemical & biological characterization), setting specification, validation, stability studies etc.
- Regulatory advice on vaccine development .
- Regulatory support for technology transfer package – review of package, gap identification etc.
- Support for pre-submission advisory meetings/ Scientific advice packages, agency meetings
Non-clinical Phase
- Guidance and review of non-clinical study protocols & reports, authoring Dossier sections
- In vitro – Functional assays, receptor binding assays, cell based assays
- In vivo – PK/PD, Comparative repeat dose toxicity, Immunogenicity (dose, dosing regimen, ROA) and Local tolerance studies, Challenge studies
Phase I
- Guidance and review of Phase I (PK/PD) clinical study protocol & report
- Support for pre-submission advisory meetings/ Scientific advice packages
Phase II (for vaccines)
- Guidance and review of Phase II clinical study protocol & report (evaluating short-term side effects & immune responses)
- Support for pre-submission advisory meetings/ Scientific advice packages
Phase III
- Guidance and review of Phase III clinical study protocol & report
- Support for pre-submission advisory meetings/ Scientific advice packages
- Authoring/ Compilation/ review of Non-clinical section of the dossier [Module 2 (2.5, 2.7), Module 5]
Regulatory strategies for global regulatory submissions of Biosimilars and Vaccines
BRICS-TM
Brazil, Russia, India, China, South Africa, Turkey and Mexico
WHO
World Health Organization
Other ROW/ EM
Rest of the world /Emerging Markets
US
United States
EU
European Union
ACCESS
Australia, Canada, United Kingdom, Singapore and Switzerland
BRICS-TM countries
End-to-end regulatory support for MAA submission in Brazil, Russia, India, South Africa, Turkey, Mexico
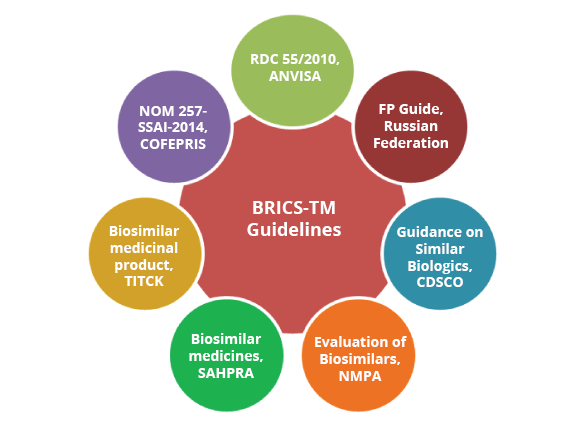
- Regulatory guidance on compliance to current regulations across BRICS-TM, gap assessment and devising regulatory strategy
- Support for dossier preparation, review and submission in BRICS-TM countries
- Evaluation of applications for registration of biological medicines, including;
- Review of dossiers for registration of biological medicines;
- Handling all matters relating to biological medicines during review of applications for registration.
- Addressing queries from regulatory agencies and supporting product registration
- Evaluation of applications for amendments to registered biological medicines
- GMP inspection and compliance support
- MA holding and life cycle management
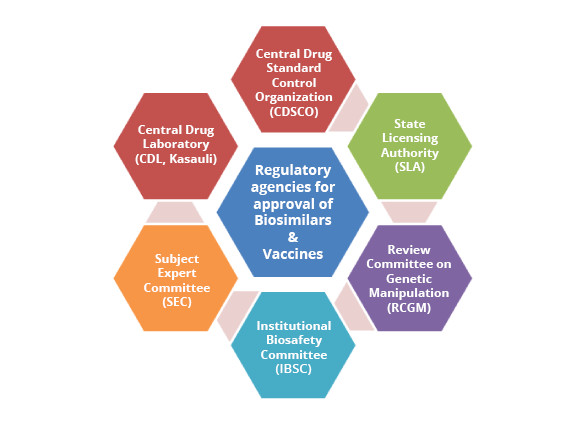
INDIA Regulatory Support
Import & Manufacture of New Drugs (CDSCO/SLA)
Registration & Import Licenses (CDSCO/ SLA)
Preclinical Protocol and Report submission & Approval (RCGM)
Clinical trial Protocol and Report Submission & Approval (CDSCO/DCGI)
Sample testing & Batch Release Certificate (CDL, Kasauli)
Marketing Authorization Application Submission & Approval (CDSCO/ DCGI)
- End-to-end regulatory support including licensure for import/export, test, examination, manufacture and approval – Form 10, 28D, 29, 46, etc.
- RCGM submissions/approval (IBK Portal) – Preparation of packages for Preclinical studies permission & approval (Form C3/C4), Submission & approval of preclinical study report (Form C5/C6)
- DCGI submissions (SUGAM portal) – Phase 1, Phase 2 and Phase 3 CTA submission package (as per New Drug Clinical Trial Rules, 2019) (Form 44)
- MAA submission support – CTD authoring/compilation, review and submission (Form 27D/28D)
- Regulatory support for filing of variations/amendments
- MA holding and lifecycle management
- Support for sample testing for biosimilar products and vaccines (CDL, Kasauli)
WHO Prequalification Programme & Other ROW/ Emerging Markets
1
Submission to WHO Prequalification programme (PQP) for Biologics
1
Submission to WHO Prequalification programme (PQP) for Biologics
2
Based on Stringent regulatory agency (SRA) approval (1) Full assessment pathway (2) Abridged assessment pathway
2
Based on Stringent regulatory agency (SRA) approval (1) Full assessment pathway (2) Abridged assessment pathway
3
GMP inspection/ Sample analysis (depends on the filing pathway)
3
GMP inspection/ Sample analysis (depends on the filing pathway)
4
Product dossier assessment by WHO
4
Product dossier assessment by WHO
5
Products assessed and placed in WHO’s list of prequalified medicines
5
Products assessed and placed in WHO’s list of prequalified medicines
6
Accelerated registration in other low-and-middle-income countries (LMICs)
6
Accelerated registration in other low-and-middle-income countries (LMICs)
7
Enables procurement by many NRAs and other UN agencies
7
Enables procurement by many NRAs and other UN agencies

Regulatory Support from Metina
- Regulatory advice and complete regulatory support for application to WHO PQP in compliance with WHO-PQP product specific guidelines & other guidance documents under WHO Technical Report Series
- WHO Pilot Procedure for Prequalification of Biotherapeutic Products: Rituximab and trastuzumab, Version 10, February 2020
- WHO Pilot Procedure for Prequalification of Biotherapeutic Products: Human Insulin, Version 10, February 2020
- Procedure for assessing the acceptability, in principles, of vaccines for purchase by United Nations agencies (2013); WHO Technical Report Series, No. 978, Annex 6.
- Support for GMP inspection/ compliance and sample testing.
- End-to-end regulatory support for filing to other ROW markets based on prequalification by WHO
Regulated Markets - US & EU
- Regulatory strategy and gap assessment for biologic/ biosimilar development for US & EU
- Regulatory support for BLA filing under 351(k) pathway of the PHS Act- authoring, review and submission
- Regulatory support for MAA submission via Centralized procedure under Article 10(4) application – authoring, review and submission
- Pre-submission guidance and support for preparation of briefing packages (BPD meetings) and scientific advice packages
- Preparation for pre-IND/ scientific advisory meetings and support for IND/IMPD package preparation and submission
- Support for GMP inspection
GMP Compliance Support for Biological Facilities

Support for EU GMP and PIC/S GMP inspection (Via NPRA, Malaysia)
GMP upgradation advice for biologics/ biosimilar manufacturing sites
Gap Analysis, Auditing and Report
Inspection Readiness Support
Preparation of GMP inspection application
Support throughout inspection, CAPA, approval & renewal
